欧盟医疗器械法规(MDR) 对医疗器械制造商提出了很多新的、更高的要求,其中包含了强化制造商的义务。
那么,作为医疗器械制造商,有哪些义务呢?本次课程将2期内容和大家进行详细说明。
欧盟授权代表
MDR Article 11
欧盟授权代表应与制造商签订委托书。
应验证制造商已经建立了EU符合性声明以及技术文件,并且实施了适当的符合性评定程序。
欧盟授权代表应保存技术文件、EU符合性声明和CE证书复印件,以备主管部门进行检查。
-如果制造商没有遵守Art. 10所规定的义务,欧盟授权代表应和制造商在同等基础上共同对有缺陷的医疗器械承担各自的法律责任。
-应检查制造商是否申请了UDI并注册。
-终止委托之后应立即通知成员国主管部门。
合规负责人
MDR Article 15
制造商必须拥有至少一名具有医疗器械领域专业知识的合规负责人。
合规负责人应确保:
按照QMS的要求对放行前的产品进行检验
建立并保持最新的技术文件和符合性声明
上市后监督
警戒系统事故报告
签署临床调查声明
提供给病人的信息和植入卡
MDR Article 18
适用范围:所有植入物
但不包括以下植入物:缝线、吻合钉、牙科矫治器、牙科填充物、牙冠、螺钉、楔子、板、丝、针、夹子和连接器等。
应包括的信息:
-用于识别产品的基本信息
-警告、注意事项等
-使用期限及必要的随访
-任何其他确保患者安全使用的信息
系统和组合包装产品
MDR Article 22/29
制造商将带有CE标识的器械与其他器械或产品一起组合,则需要起草相应的声明,还应按照指定发行实体的规则,向系统或组合包装产品分配基本UDI – DI,并将其与在附录VI第B部分中定义的相关系统或组合包装产品的其他核心数据要素一并提交至UDI数据库。
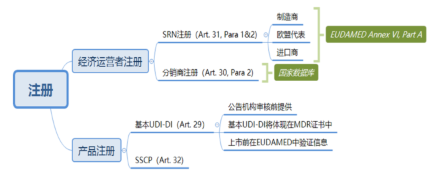
注册/SRN
安全和临床性能总结
MDR Article 32
适用于:III类产品和可植入产品
制造商的职责:
-负责起草SSCP
-SSCP的草稿应是CE技术文件的一部分,根据符合性评估程序提交给公告机构进行确认
-制造商应在标签或说明书中告知可以获得SSCP的地方
-至少每年更新一次
SSCP的要求:应让预期的使用者及(若相关)患者明白,并应通过Eudamed向公众开放
联系我们
如需了解更多信息,请与我们联系:
电话:021-80317636
邮箱:info@landlink-healthcare.com


