这是一个大话题。在我们日常工作中,常会碰到这种类型的咨询,产品变更的类型多种多样,变更的原因也五花八门,但核心的疑问只有一个:变更后能否不作新的510(k)申请。
FDA专门关于这个话题发布了一份指南:Deciding When to Submit a 510(k) for a Change to an Existing Device。这份指南有77页,小编建议在通读一遍的基础上,作为工具书使用,因为该指南针对了多种变更的情况,要全部透彻消化很费劲且没必要。
指南主要概括了以下情况的变更:
Labeling Changes
Technology, Engineering, and Performance Changes
Materials Changes
Technology, Engineering, Performance, and Materials Changes for In Vitro Diagnostic Devices(只针对IVD产品)
每个章节都有对应的判断流程图,同时也有概要的主体判断流程图(下文会提及)。可以看出,排版上也非常适合作为工具书使用。
核心原则
FDA与国内注册不同,不存在注册变更的行政操作(更改地址这些事情,直接在注册列名系统改动即可)。产品变更只有需要新的递交510(k)申请或不需要递交的分别,会否触发新的510(k)申请,核心判断原则是:变更是否对现有安全性或有效性产生明显变化。变化有两方面,正面和负面变化,但不管哪方面变化,只要是与产品现状产生明显安全性或有效性差异,都需要递交新的510(k)申请。
已注册产品的变更,通常分为两种类型,有意变更与无意变更。有意变更是指特意对产品安全性或有效性改善的变更,会发生在上市后收到客户投诉或产品召回等情况。无意变更是日常工作中最常见的情形,制造商无意改变产品现有安全性或有效性,但因为供应商改变、生产设备升级等原因,而对产品有效性或安全性不可避免的产生了影响。对于有意变更,基本上不用多想,准备新的510(k)申请就完事了。但是对于无意变更,则需要进行下文会提到Risk-Based Assessments的判断,来决定是否需要递交新的510(k)申请。
下图是主体判断流程图,属于哪个变更,则要回到对应章节再进行细判断。
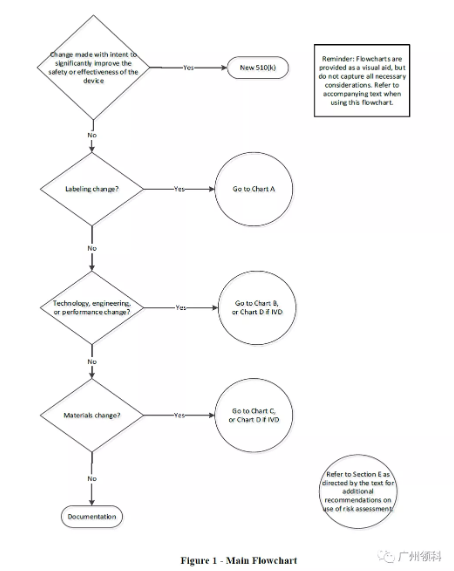
有些读者可能会奇怪,为什么没有生产制造过程变更的章节?并不是说生产制造过程变更不需要考虑,相反地,是因为FDA认为生产制造过程的变更最终会导致的是上文提及A、B/D、C的变更,而且影响可能不止一个方面,所以引导制造商往生产制造过程变更造成的最终影响方向考虑,而不单独列出来局限大家判断。
是不是最后判断为不需要递交新的510(k)申请就完事呢?不是的。根据指南要求,制造商需要将整个判断过程记录下来,作为体系文件保存,以备飞检抽查之用。
Risk-Based Assessments
无意变更是否要递交新的510(k)申请,不是简单一句需要或不需要就完事的,是要基于Risk-Based Assessments的判断。这个风险判断与产品上市前的Benefit-Risk Assessment是不一样的,基本上与获益无关,就是基于安全性是否有降低的判断。安全性降低体现在两方面,一是产生新的hazard或hazard situation,二是将现有的risk的可能性或严重性提高。制造商要分别对这两方面进行判断,要是最后结论是风险的新变化是在原有可接受范围,那就可以不递交新的510(k)申请,如果是在不可接受范围,那么则需要采取风险控制手段并且进行新的510(k)申请。
有效性
Risk-Based Assessments主要针对的是对产品安全性的评估,但除此以外,还有有效性的评估。很多方面的变更都会影响产品有效性,有时候甚至是为了降低产品安全性风险而采取的控制手段,也会对产品有效性有负面影响。
如何判断产品有效性能否保持现状,最直接的方法就是进行性能验证和确认,按照原来的测试方法,对成品进行测试。如果测试结果结果与过去保持一致,那就证明有效性能保持;但是如果出现意料之外的结果(不合格),制造商就要进行分析了,要么是过去的测试方法或判断标准已经不适用于变更后的产品,要么是产品的有效性真的降低了。不论是那种原因,只要出现使用原有测试方法与判定标准,变更后的产品得出意料之外的结果,都需要递交新的510(k)申请。
我换了测试方法后,是能通过测试的,那是不是不需要递交新的510(k)。抱歉哦,FDA已经在指南中特意明确了这种类型的情况是不被允许的,希望大家就不要抱有侥幸心态了。
原文: If different verification and/or validation test methods or
acceptance criteria are necessary to produce the expected results, it is likely that the change could significantly affect safety or effectiveness and thus submission of a new510(k) is likely required.
一定要注意的是: 只要Risk-Based Assessments后,是明确会对现有安全性或有效性产生明显变化,哪怕是有效性测试通过,都是需要递交新的510(k)申请。
递交新510(k)申请的准备工作
如果明确了是需要递交新的510(k)申请,除了要列出导致要递交新510(k)申请的变更外,其他的没对现有安全性或有效性造成明显差异的变更也要列上(有些产品变更不止一处)。
此外,大家一定一定要格外注意:以上所有内容都是针对已注册产品变更是否会触发新的510(k)申请的,并没有说新的510(k)申请就能继续使用原来的比对器械啊。一定要注意比较变更后的产品是否与原有的比对器械匹配,不然会得到NSE的结果哦~
实战例子
说了那么多,直接用一个小编刚接触到的例子和大家实战演绎。客户因为灭菌设备的改变,由钴60灭菌改成电子束灭菌,但仍然是采用辐照灭菌的方式,只是具体方法不同了。根据主体判断流程图了解,这不是有意要改变产品安全性或有效性的变更,进入章节B环节。

在章节B的判断流程图中,可以看出核心问题就是B3.1与B3.2
Question B3.1 Is it a change to an “established category B” or “novel” sterilization method, does the change lower the sterility assurance level, or is it a change to how the device is provided?
小编注:FDA对灭菌方式是有分类的,分为A组、B组与novel组别,风险程度是A组<B组<novel,常见的辐照灭菌、EO灭菌、湿热灭菌都是处于A组,当产品灭菌方式是由A组往其他组别改变时,肯定需要递交新的510(k)申请。
在这个客户的实际情况上,以上三个问题的答案都是否定的,可以进入B3.2。这里啰嗦一下,关于“or is it a change to how the device is provided”这个问题,其实在口罩FXX注册中也常遇见。FDA口罩FXX注册多以非灭菌方式,但是制造商很多时候为了方便,都会以灭菌方式交付(因为CE大多是1s类),虽然看上去更干净,但这种变更是需要递交新的510(k)申请,因为交付产品从“非灭菌”改成“灭菌”是会对现有安全性或有效性产生明显差异。
Question B3.2 Could the change significantly affect the performance or biocompatibility of the device?
这个是个非常好的提问。也是小编与客户讨论的重点。不是单纯说完成了灭菌验证、无菌测试,就证明无明显差异了。还需要做与过去一致的性能测试、包装运输测试、加速老化测试等,证明产品在规定的生命周期中,是能保持与过往一致的有效性,才真正是不需要递交新的510(k)申请。
以上就是今天的分享了,内容有些多。如果各位制造商产品FDA注册后变更,拿捏不准是否需要递交新的510(k)申请的话,欢迎与我们联系,相信我们技术团队会给您一个满意的方案
参考来源:
[1] Deciding When to Submit a 510(k) for a Change to an Existing Device
[2] Submission and Review of Sterility Information in Premarket Notification (510(k)) Submissions for Devices Labeled as Sterile

联系我们
如需了解更多信息,请与我们联系:
电话:021-80317636
邮箱:info@landlink-healthcare.com


